
Diagnostic Expertise
Full-service execution from protocol design through regulatory submission.
For sponsors
Studybox partners with IVD sponsors as a strategic advisor and hands-on executor. We bring the diagnostic expertise, the right sites, and the operational infrastructure so your study moves faster, costs less, and gets done right.
50+
Regulatory clearances
30+
Years of experience
100%
IVD exclusive focus
100+
Pre-Qualified Sites
Clinical research services
Every study we run is an in vitro diagnostic study: from protocol to FDA response, one team, one point of contact, zero handoffs.
Full-service execution from protocol design through regulatory submission.
Pre-established site relationships matched to your IUO profile.
Site network spanning demographics, geographies, and clinical settings.
Streamlined pathways under a single point of expertise.
Our process
We design or refine your protocol to meet FDA requirements and maximize site feasibility.
We map the right regulatory pathway: 510(k), CLIA Waiver, Dual Submission, Emergency Use Authorization, De Novo, Reproducibility and Precision studies.
We match your IUO profile to sites across our established network for rapid activation.
Enrollment, supply management, and site operations run under our direct oversight.
Live dashboards, data review, QA, and deviation management throughout the study.
Study report, FDA response support, and clearance readiness delivered on time.
Site Network
A geographically diverse clinical research site network spanning point-of-care, urgent care, and home-use settings, pre-qualified, saving you time and money.
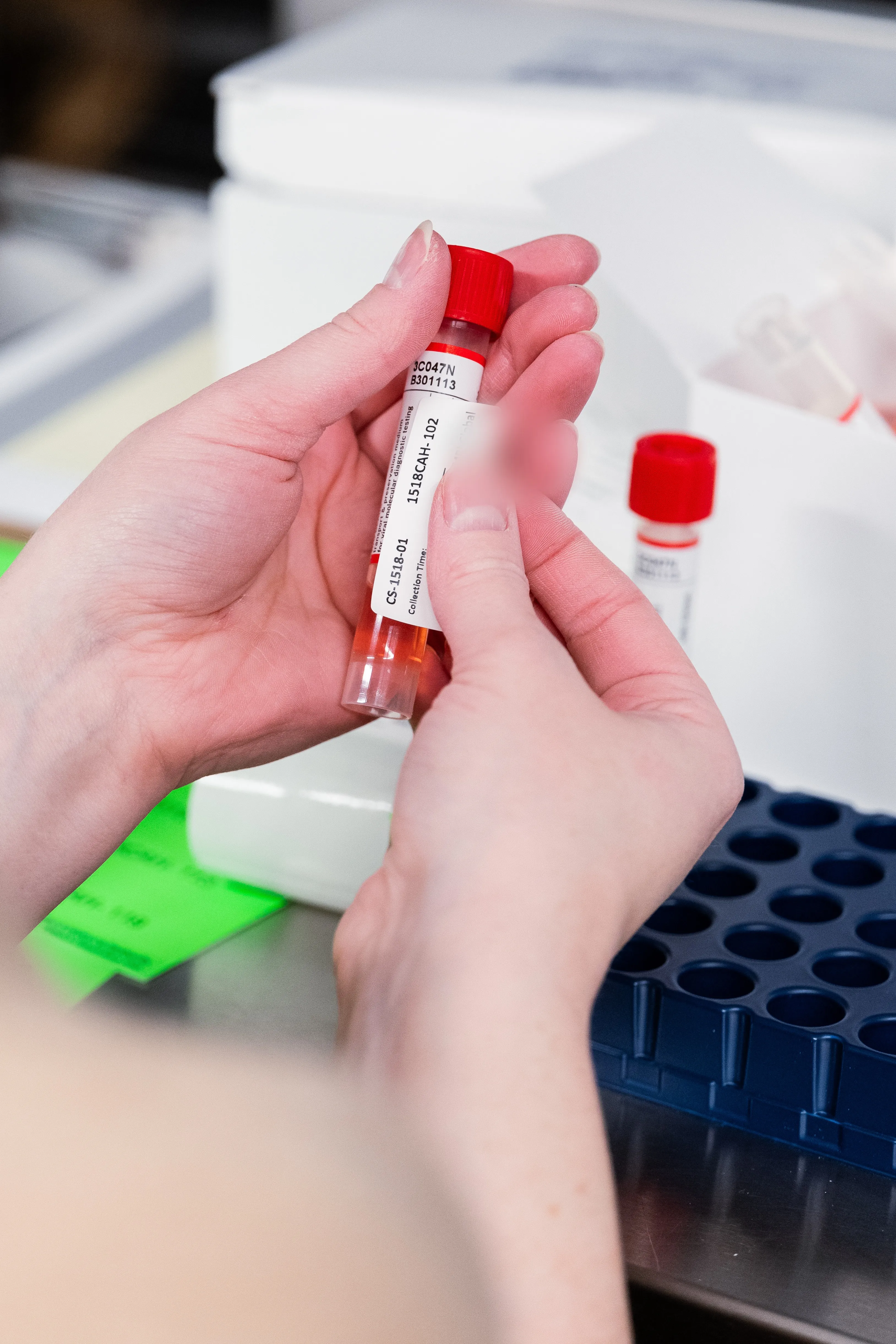
Specimen collection
Launch new collections in as little as one week, with no study startup required.
Studybox maintains a pre-approved IRB protocol covering the most common specimen types, so sponsors can access fresh, prospectively collected specimens without the time or cost of initiating a dedicated study. Data capture is fully customizable to your needs, including demographics, concomitant medications, symptom onset, symptom profile, and more.
FAQ
Because our sites are pre-qualified for IVD protocols, we can typically activate within approximately 4 weeks of study initiation, compared to the 3 to 6 month industry average.
We support 510(k), CLIA Waiver, Dual Submission, Emergency Use Authorization, De Novo, reproducibility and precision studies.
Our regulatory team reviews protocols before study start to minimize amendment risk. When amendments do occur, we manage the IRB/Ethic's Committee communication and site updates end-to-end.
Yes. We work with international sponsors seeking FDA clearance for their IVD products, providing US-based site networks and regulatory expertise.
Timeline depends on study complexity, enrollment requirements, and regulatory pathway. We provide detailed project timelines at study initiation and update them weekly throughout execution.
Start the conversation
Tell us about your program and we will follow up within one business day.
Prefer to email?
[email protected]